-
-
-
Jakarta, Indonesia
Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
The "Lazy Genius" of Bacteria: Unlocking the Molecular Pathways of Morbus Hansen (Leprosy)
-
 Post By
Post By -
Published
March 12, 2026
A Pathogen That Thinks in Phases
Mycobacterium leprae is, by almost any metric, a remarkable organism. It possesses the smallest functional genome of any known mycobacterium, having shed roughly half its ancestral genetic material through reductive evolution over millions of years of obligate intracellular parasitism. It cannot be cultured on any artificial medium. Its generation time of approximately thirteen days is the slowest of any known human bacterial pathogen. And yet, despite — or more precisely, because of — this apparent biological minimalism, it has perfected a three-phase molecular strategy for establishing itself within the human nervous system, reprogramming the very cells designed to maintain nerve integrity, and manipulating the host immune response in ways that allow it to persist for years to decades before the full clinical consequences of its presence become apparent.
Morbus Hansen — leprosy — is a disease that has accompanied humanity for at least four thousand years, documented in ancient Indian, Chinese, and Egyptian texts, and carrying a cultural stigma so profound that it has shaped the social history of societies on every inhabited continent. Yet despite this long coexistence, the molecular mechanisms by which M. leprae achieves its predilection for peripheral nerve tissue, its extraordinary survival within host cells, and its ability to generate a clinical spectrum ranging from self-limiting paucibacillary disease to progressive multibacillary dissemination, remained incompletely understood until the late twentieth and early twenty-first centuries.
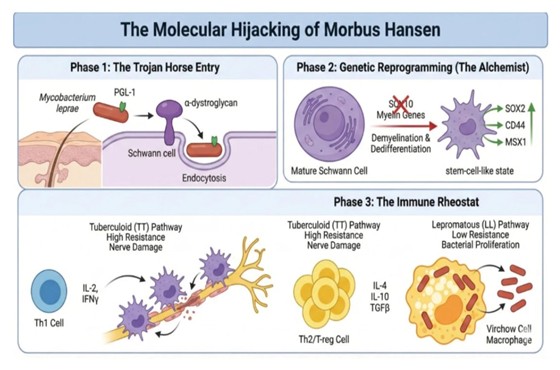
The figure above condenses this understanding into a three-phase framework: the Trojan horse entry into Schwann cells, the genetic reprogramming of those cells into a dedifferentiated, stem-cell-like state that serves the bacterium's replicative needs, and the immunological rheostat whose polarization determines whether a patient develops the high-resistance tuberculoid form or the low-resistance lepromatous form of the disease. Each phase reveals a different facet of M. leprae's strategic sophistication — and each phase offers potential therapeutic intervention points that modern leprosy research is beginning to exploit.
Morbus Hansen — leprosy — is a disease that has accompanied humanity for at least four thousand years, documented in ancient Indian, Chinese, and Egyptian texts, and carrying a cultural stigma so profound that it has shaped the social history of societies on every inhabited continent. Yet despite this long coexistence, the molecular mechanisms by which M. leprae achieves its predilection for peripheral nerve tissue, its extraordinary survival within host cells, and its ability to generate a clinical spectrum ranging from self-limiting paucibacillary disease to progressive multibacillary dissemination, remained incompletely understood until the late twentieth and early twenty-first centuries.
The figure above condenses this understanding into a three-phase framework: the Trojan horse entry into Schwann cells, the genetic reprogramming of those cells into a dedifferentiated, stem-cell-like state that serves the bacterium's replicative needs, and the immunological rheostat whose polarization determines whether a patient develops the high-resistance tuberculoid form or the low-resistance lepromatous form of the disease. Each phase reveals a different facet of M. leprae's strategic sophistication — and each phase offers potential therapeutic intervention points that modern leprosy research is beginning to exploit.
The Predilection for Peripheral Nerve: Why Schwann Cells?
Before examining the molecular details of cellular entry, it is worth interrogating the more fundamental question: why the peripheral nervous system at all? What biological feature of the Schwann cell — the myelinating and non-myelinating glial cell of the peripheral nerve — makes it the preferred intracellular refuge of M. leprae when other intracellular pathogens more commonly target macrophages, dendritic cells, or epithelial cells?
Several complementary explanations have been proposed, and the weight of evidence supports a convergence of factors. First, the peripheral nerve microenvironment offers relative immunological privilege: the blood-nerve barrier, analogous to the blood-brain barrier of the central nervous system, limits the access of circulating immune cells and antibodies to the endoneurium, creating a sanctuary within which intracellular bacteria can persist with reduced immunological surveillance. Second, Schwann cells express the specific surface receptors that M. leprae has evolved to target — a molecular compatibility that appears to represent the product of millions of years of host-pathogen co-evolution. Third, the biological state to which M. leprae drives Schwann cells — the dedifferentiated, stem-cell-like phenotype described in Phase 2 — creates an intracellular environment that is demonstrably more permissive to bacterial survival and replication than the differentiated mature Schwann cell it replaces.
The clinical consequence of this predilection is the feature that makes leprosy unique among bacterial infections: the cardinal sign of peripheral neuropathy. While skin lesions are often the first manifestation that brings patients to medical attention, it is the destruction of peripheral nerves — the thickened, tender nerve trunks, the sensory loss that renders patients unable to perceive injurious stimuli, the motor dysfunction that leads to the characteristic hand and foot deformities — that generates the lifelong disability associated with this disease. Understanding how M. leprae enters, reprograms, and eventually destroys peripheral nerve is therefore not merely academic; it is the mechanistic foundation upon which neuroprotective therapeutic strategies must be built.
Several complementary explanations have been proposed, and the weight of evidence supports a convergence of factors. First, the peripheral nerve microenvironment offers relative immunological privilege: the blood-nerve barrier, analogous to the blood-brain barrier of the central nervous system, limits the access of circulating immune cells and antibodies to the endoneurium, creating a sanctuary within which intracellular bacteria can persist with reduced immunological surveillance. Second, Schwann cells express the specific surface receptors that M. leprae has evolved to target — a molecular compatibility that appears to represent the product of millions of years of host-pathogen co-evolution. Third, the biological state to which M. leprae drives Schwann cells — the dedifferentiated, stem-cell-like phenotype described in Phase 2 — creates an intracellular environment that is demonstrably more permissive to bacterial survival and replication than the differentiated mature Schwann cell it replaces.
The clinical consequence of this predilection is the feature that makes leprosy unique among bacterial infections: the cardinal sign of peripheral neuropathy. While skin lesions are often the first manifestation that brings patients to medical attention, it is the destruction of peripheral nerves — the thickened, tender nerve trunks, the sensory loss that renders patients unable to perceive injurious stimuli, the motor dysfunction that leads to the characteristic hand and foot deformities — that generates the lifelong disability associated with this disease. Understanding how M. leprae enters, reprograms, and eventually destroys peripheral nerve is therefore not merely academic; it is the mechanistic foundation upon which neuroprotective therapeutic strategies must be built.
Phase 1: The Trojan Horse Entry
PGL-1: The Bacterium's Master Key
The molecular mechanism of M. leprae's entry into Schwann cells is among the most precisely characterized host-pathogen interactions in microbiology. The critical ligand on the bacterial surface is phenolic glycolipid-1 — PGL-1 — a unique trisaccharide-containing glycolipid that is specific to M. leprae and absent from all other mycobacterial species, including Mycobacterium tuberculosis. This molecular specificity is significant: PGL-1 is not merely a surface antigen but a molecular key evolved for a single lock, and its identification as the primary adhesin for Schwann cell invasion explains at the biochemical level why M. leprae — and only M. leprae — generates peripheral neuropathy as the defining pathological signature of its infection.
The receptor to which PGL-1 binds on the Schwann cell surface is alpha-dystroglycan — a component of the dystrophin-associated glycoprotein complex that, under normal physiological circumstances, mediates the attachment of Schwann cells to the extracellular matrix component laminin-2 of the peripheral nerve basal lamina. Alpha-dystroglycan is a heavily glycosylated protein whose carbohydrate modifications are essential both for its normal laminin-binding function and for its susceptibility to PGL-1 binding. The specific sugar residues that M. leprae targets are O-mannosyl glycans that decorate the mucin-like domain of alpha-dystroglycan — the same glycan modifications that are disrupted in a group of congenital muscular dystrophies collectively termed dystroglycanopathies, and whose biological significance was only fully appreciated after the discovery of their role in mycobacterial invasion.
Endocytosis: Tricking the Cell into Opening Its Doors
When PGL-1 binds alpha-dystroglycan, it does not merely achieve surface attachment — it actively subverts the Schwann cell's intracellular signaling to trigger its own internalization. The PGL-1-alpha-dystroglycan interaction recruits ErbB2/ErbB3 receptor tyrosine kinases to the contact site and activates downstream signaling cascades including the PI3K-Akt pathway and focal adhesion kinase, collectively initiating macropinocytosis and phagocytic cup formation around the bacterium. The Schwann cell, deceived by the molecular mimicry of a legitimate matrix-adhesion signal, engulfs the bacterium through a process of receptor-mediated endocytosis that delivers it into an intracellular vesicular compartment.
Once internalized, M. leprae faces the same challenge that confronts all intracellular mycobacteria: survival within the phagosomal compartment. Like its relative M. tuberculosis, M. leprae employs mechanisms to inhibit phagosome-lysosome fusion, preventing the delivery of its endosomal vacuole to the acidified, hydrolase-rich lysosomal environment that would otherwise destroy it. Within the resulting phagosome — maintained at a relatively neutral pH and protected from lysosomal enzymes — M. leprae replicates at its characteristically slow pace, shielded from both extracellular immune effectors and the intracellular killing mechanisms that the Schwann cell, as a non-professional phagocyte, is in any case poorly equipped to deploy.
The Trojan horse metaphor employed in Figure 1 is apt in a precise molecular sense: M. leprae does not breach the Schwann cell by force but by presenting a surface molecule that is recognized as belonging to a normal biological interaction — and is therefore welcomed inside. The strategy bypasses the cell's defensive reflexes entirely, because no defensive reflex is triggered. The bacterium has not forced a door; it has been invited through one.
The molecular mechanism of M. leprae's entry into Schwann cells is among the most precisely characterized host-pathogen interactions in microbiology. The critical ligand on the bacterial surface is phenolic glycolipid-1 — PGL-1 — a unique trisaccharide-containing glycolipid that is specific to M. leprae and absent from all other mycobacterial species, including Mycobacterium tuberculosis. This molecular specificity is significant: PGL-1 is not merely a surface antigen but a molecular key evolved for a single lock, and its identification as the primary adhesin for Schwann cell invasion explains at the biochemical level why M. leprae — and only M. leprae — generates peripheral neuropathy as the defining pathological signature of its infection.
The receptor to which PGL-1 binds on the Schwann cell surface is alpha-dystroglycan — a component of the dystrophin-associated glycoprotein complex that, under normal physiological circumstances, mediates the attachment of Schwann cells to the extracellular matrix component laminin-2 of the peripheral nerve basal lamina. Alpha-dystroglycan is a heavily glycosylated protein whose carbohydrate modifications are essential both for its normal laminin-binding function and for its susceptibility to PGL-1 binding. The specific sugar residues that M. leprae targets are O-mannosyl glycans that decorate the mucin-like domain of alpha-dystroglycan — the same glycan modifications that are disrupted in a group of congenital muscular dystrophies collectively termed dystroglycanopathies, and whose biological significance was only fully appreciated after the discovery of their role in mycobacterial invasion.
Endocytosis: Tricking the Cell into Opening Its Doors
When PGL-1 binds alpha-dystroglycan, it does not merely achieve surface attachment — it actively subverts the Schwann cell's intracellular signaling to trigger its own internalization. The PGL-1-alpha-dystroglycan interaction recruits ErbB2/ErbB3 receptor tyrosine kinases to the contact site and activates downstream signaling cascades including the PI3K-Akt pathway and focal adhesion kinase, collectively initiating macropinocytosis and phagocytic cup formation around the bacterium. The Schwann cell, deceived by the molecular mimicry of a legitimate matrix-adhesion signal, engulfs the bacterium through a process of receptor-mediated endocytosis that delivers it into an intracellular vesicular compartment.
Once internalized, M. leprae faces the same challenge that confronts all intracellular mycobacteria: survival within the phagosomal compartment. Like its relative M. tuberculosis, M. leprae employs mechanisms to inhibit phagosome-lysosome fusion, preventing the delivery of its endosomal vacuole to the acidified, hydrolase-rich lysosomal environment that would otherwise destroy it. Within the resulting phagosome — maintained at a relatively neutral pH and protected from lysosomal enzymes — M. leprae replicates at its characteristically slow pace, shielded from both extracellular immune effectors and the intracellular killing mechanisms that the Schwann cell, as a non-professional phagocyte, is in any case poorly equipped to deploy.
The Trojan horse metaphor employed in Figure 1 is apt in a precise molecular sense: M. leprae does not breach the Schwann cell by force but by presenting a surface molecule that is recognized as belonging to a normal biological interaction — and is therefore welcomed inside. The strategy bypasses the cell's defensive reflexes entirely, because no defensive reflex is triggered. The bacterium has not forced a door; it has been invited through one.
Phase 2: Genetic Reprogramming — The Alchemist Within
SOX10 Suppression and the Dismantling of Schwann Cell Identity
Having established itself within the Schwann cell, M. leprae does not simply reside passively in its intracellular niche. It actively rewires the transcriptional program of its host cell — a feat of molecular manipulation that serves the bacterium's survival interests with a sophistication that rivals the most complex viral cellular reprogramming events. The central target of this reprogramming is SOX10, a transcription factor of the SRY-related HMG-box family that is essential for the maintenance of the mature Schwann cell phenotype.
In the differentiated, myelinating Schwann cell, SOX10 drives the expression of the myelin gene program — including myelin protein zero (MPZ), myelin basic protein (MBP), peripheral myelin protein 22 (PMP22), and the lipid biosynthesis enzymes that generate the myelin sheath. This transcriptional program is what defines the Schwann cell as a myelinating cell and what makes it capable of insulating axons with the multilamellar myelin membrane that enables saltatory conduction and protects axonal integrity. Mycobacterium leprae suppresses SOX10 activity through mechanisms that involve both direct epigenetic modification of the SOX10 promoter and the downregulation of upstream activators of SOX10 expression, disabling the master regulator of Schwann cell differentiated identity.
The consequence of SOX10 suppression is demyelination — the loss of the myelin sheath that surrounds peripheral axons — and cellular dedifferentiation, the process by which the mature, specialized Schwann cell loses its differentiated phenotype and reverts toward a more primitive, multipotent state. Clinically, this demyelination is the direct substrate of the nerve conduction slowing and sensory loss that are the earliest neurological manifestations of leprosy, detectable on nerve conduction studies before any anatomically visible nerve damage has occurred.
The Stem-Cell-Like State: Turning the Clock Back for Bacterial Benefit
The dedifferentiation of M. leprae-infected Schwann cells does not produce a biologically inert or simply damaged cell. It produces a cell that actively upregulates a suite of transcription factors and surface markers associated with neural crest stem cells — the embryonic progenitor population from which Schwann cells originally derive during development. The key markers of this reprogrammed state include SOX2, the pluripotency transcription factor that drives neural stem cell self-renewal; CD44, a cell adhesion molecule and receptor for hyaluronic acid that marks mesenchymal stem cell populations; and MSX1, a homeobox transcription factor involved in neural crest cell multipotency.
This reprogrammed, stem-cell-like Schwann cell is, from the bacterium's perspective, a far more hospitable host than its differentiated precursor. The stem-cell-like state is associated with enhanced migratory capacity — a property that M. leprae exploits to disseminate from peripheral nerve to skin and to spread between nerve fascicles within the same nerve trunk. The reprogrammed cells are also more resistant to apoptosis, extending their lifespan and providing the bacterium with a longer-lived intracellular reservoir. And the stem-cell-like phenotype appears to create a metabolic environment more supportive of mycobacterial lipid synthesis and slow replication than the highly specialized metabolic profile of the mature myelinating Schwann cell.
The pathological elegance of this strategy is worth pausing to appreciate. M. leprae, a bacterium so metabolically reduced that it cannot synthesize many of its own amino acids, lipids, and nucleotides — instead depending entirely on host cell biosynthesis for these essential building blocks — has evolved the capacity to reprogram a terminally differentiated adult cell back into a stem-cell-like state that is metabolically and phenotypically optimized for bacterial survival. It is, in the most literal sense of the term, an alchemical transformation: the bacterium converts the mature Schwann cell into something fundamentally different, and something that serves its own purposes far better than the original.
The broader implications of this reprogramming for neural biology extend beyond leprosy itself. The discovery that a bacterium can reproducibly dedifferentiate adult peripheral glia into neural crest-like stem cells has stimulated significant interest in whether M. leprae-derived or M. leprae-inspired molecular tools could be harnessed for regenerative medicine applications — an area of active investigation that represents one of the more unexpected scientific offshoots of leprosy research
Having established itself within the Schwann cell, M. leprae does not simply reside passively in its intracellular niche. It actively rewires the transcriptional program of its host cell — a feat of molecular manipulation that serves the bacterium's survival interests with a sophistication that rivals the most complex viral cellular reprogramming events. The central target of this reprogramming is SOX10, a transcription factor of the SRY-related HMG-box family that is essential for the maintenance of the mature Schwann cell phenotype.
In the differentiated, myelinating Schwann cell, SOX10 drives the expression of the myelin gene program — including myelin protein zero (MPZ), myelin basic protein (MBP), peripheral myelin protein 22 (PMP22), and the lipid biosynthesis enzymes that generate the myelin sheath. This transcriptional program is what defines the Schwann cell as a myelinating cell and what makes it capable of insulating axons with the multilamellar myelin membrane that enables saltatory conduction and protects axonal integrity. Mycobacterium leprae suppresses SOX10 activity through mechanisms that involve both direct epigenetic modification of the SOX10 promoter and the downregulation of upstream activators of SOX10 expression, disabling the master regulator of Schwann cell differentiated identity.
The consequence of SOX10 suppression is demyelination — the loss of the myelin sheath that surrounds peripheral axons — and cellular dedifferentiation, the process by which the mature, specialized Schwann cell loses its differentiated phenotype and reverts toward a more primitive, multipotent state. Clinically, this demyelination is the direct substrate of the nerve conduction slowing and sensory loss that are the earliest neurological manifestations of leprosy, detectable on nerve conduction studies before any anatomically visible nerve damage has occurred.
The Stem-Cell-Like State: Turning the Clock Back for Bacterial Benefit
The dedifferentiation of M. leprae-infected Schwann cells does not produce a biologically inert or simply damaged cell. It produces a cell that actively upregulates a suite of transcription factors and surface markers associated with neural crest stem cells — the embryonic progenitor population from which Schwann cells originally derive during development. The key markers of this reprogrammed state include SOX2, the pluripotency transcription factor that drives neural stem cell self-renewal; CD44, a cell adhesion molecule and receptor for hyaluronic acid that marks mesenchymal stem cell populations; and MSX1, a homeobox transcription factor involved in neural crest cell multipotency.
This reprogrammed, stem-cell-like Schwann cell is, from the bacterium's perspective, a far more hospitable host than its differentiated precursor. The stem-cell-like state is associated with enhanced migratory capacity — a property that M. leprae exploits to disseminate from peripheral nerve to skin and to spread between nerve fascicles within the same nerve trunk. The reprogrammed cells are also more resistant to apoptosis, extending their lifespan and providing the bacterium with a longer-lived intracellular reservoir. And the stem-cell-like phenotype appears to create a metabolic environment more supportive of mycobacterial lipid synthesis and slow replication than the highly specialized metabolic profile of the mature myelinating Schwann cell.
The pathological elegance of this strategy is worth pausing to appreciate. M. leprae, a bacterium so metabolically reduced that it cannot synthesize many of its own amino acids, lipids, and nucleotides — instead depending entirely on host cell biosynthesis for these essential building blocks — has evolved the capacity to reprogram a terminally differentiated adult cell back into a stem-cell-like state that is metabolically and phenotypically optimized for bacterial survival. It is, in the most literal sense of the term, an alchemical transformation: the bacterium converts the mature Schwann cell into something fundamentally different, and something that serves its own purposes far better than the original.
The broader implications of this reprogramming for neural biology extend beyond leprosy itself. The discovery that a bacterium can reproducibly dedifferentiate adult peripheral glia into neural crest-like stem cells has stimulated significant interest in whether M. leprae-derived or M. leprae-inspired molecular tools could be harnessed for regenerative medicine applications — an area of active investigation that represents one of the more unexpected scientific offshoots of leprosy research
Phase 3: The Immune Rheostat — Two Diseases from One Bacterium
The clinical spectrum of leprosy — from the paucibacillary, high-resistance tuberculoid pole to the multibacillary, low-resistance lepromatous pole — is one of the most instructive natural experiments in human immunology. No other infectious disease demonstrates so clearly, in such clinically discrete forms, the consequences of polarized T-helper cell responses for the balance between bacterial control and tissue damage. The immune rheostat that determines a patient's position on this spectrum is not set at the moment of infection but is shaped over time by genetic predispositions, prior immune experiences, and the evolving molecular dialogue between the bacterium, the infected Schwann cell, and the surrounding immune microenvironment.
The Tuberculoid Pathway: Resistance at a Price
At the tuberculoid pole, the adaptive immune response is dominated by Th1 CD4-positive T lymphocytes that recognize mycobacterial antigens presented by macrophages and dendritic cells in the context of MHC class II molecules. These Th1 cells secrete the signature cytokines of the tuberculoid response: interleukin-2, which drives T-cell proliferation and sustains the clonal expansion of antigen-specific effector cells, and interferon-gamma, the quintessential macrophage-activating cytokine that induces the production of reactive oxygen and nitrogen intermediates, upregulates antigen presentation, and promotes the granulomatous cellular architecture that is the primary mechanism of mycobacterial containment.
The clinical consequence of this vigorous Th1 response is a disease that is bacteriologically contained but immunopathologically destructive. In tuberculoid leprosy, the mycobacterial burden is low — sometimes to the point where acid-fast bacilli cannot be detected even on multiple skin slit smears — and the risk of transmission is correspondingly reduced. But the intensely activated macrophages and lymphocytes that accomplish this bacterial suppression do so at the cost of collateral damage to the Schwann cells, axons, and connective tissue of the affected nerve. Nerve trunks in tuberculoid leprosy are thickened, tender, and surrounded by caseating or non-caseating granulomas that physically compress neural structures, disrupt blood supply, and ultimately destroy nerve fascicles with irreversible functional consequences.
Tuberculoid leprosy characteristically affects only one or a few nerve trunks and produces few, well-defined, hypopigmented skin lesions with clearly demarcated borders and complete loss of sensation within the lesion. This geographical containment reflects the localized and effective immunological barrier that the Th1 response constructs around the site of infection. The disease is self-limiting in many patients, capable of resolution without treatment in those with particularly robust cell-mediated immunity — though treatment remains essential to prevent progressive nerve damage during the active inflammatory phase.
The Lepromatous Pathway: Bacterial Triumph Through Immune Evasion
At the lepromatous pole, the immune balance has been tipped decisively in the bacterium's favor. The antigen-specific Th1 response is selectively anergic — patients with lepromatous leprosy fail to mount a delayed-type hypersensitivity reaction to intradermal lepromin, demonstrating that the specific immunological recognition of M. leprae antigens, rather than general immune competence, has been impaired. This anergy is actively maintained by a regulatory immune environment dominated by Th2 cells and regulatory T cells secreting immunosuppressive cytokines.
Interleukin-4, secreted by Th2 cells, redirects the immune response away from cell-mediated cytotoxicity and toward humoral immunity — a response that is relatively ineffective against an intracellular pathogen. Interleukin-10, produced by both Th2 cells and regulatory T cells, is a potent inhibitor of macrophage activation and Th1 cytokine production, directly antagonizing the interferon-gamma signaling that would otherwise activate mycobactericidal mechanisms. Transforming growth factor-beta, the signature cytokine of regulatory T-cell immunosuppression, further restrains the effector T-cell response and promotes tolerance to mycobacterial antigens. Together, these cytokines create an immunological environment in which M. leprae can proliferate essentially unchecked.
The cellular consequence of this permissive immune environment is the formation of Virchow cells — the foamy, lipid-laden macrophages that are the histopathological signature of lepromatous leprosy and the direct structural correlate of massive bacterial proliferation. Virchow cells are macrophages that have attempted and failed to kill internalized M. leprae, instead becoming overwhelmed with replicating bacteria that fill their cytoplasm with lipid-rich vacuoles. The lipid content of these vacuoles reflects both the bacterium's own lipid-rich cell wall and the accumulation of host cell lipids that M. leprae co-opts as energy substrates and membrane building materials. In lepromatous leprosy, Virchow cells accumulate in enormous numbers throughout the dermis, in peripheral nerve, in nasal mucosa, in the testes, and in other cooler body sites that provide the slightly below-body-temperature microenvironment that M. leprae preferentially inhabits.
The clinical picture of lepromatous leprosy reflects this bacteriologically permissive state. Skin lesions are numerous, poorly defined, symmetrically distributed, and filled with bacilli — each lesion a macroscopic manifestation of a site of uncontrolled mycobacterial replication. Peripheral nerve involvement is diffuse and bilateral, affecting multiple nerve trunks symmetrically in a stocking-and-glove distribution that contrasts sharply with the asymmetric, focal neuropathy of tuberculoid disease. The eyebrows, earlobes, and nasal bridge develop the characteristic leonine facies of advanced lepromatous disease as dermal macrophage infiltration thickens and distorts the facial skin. Nasal septal perforation, from direct mycobacterial destruction of the nasal mucosa and cartilage, can produce the saddle-nose deformity that is among the most historically recognizable stigmata of the disease.
The Ridley-Jopling Spectrum: Positions on the Dial
Between the tuberculoid and lepromatous poles lies a continuous immunological spectrum that the Ridley-Jopling classification system divides into five positions: tuberculoid (TT), borderline tuberculoid (BT), mid-borderline (BB), borderline lepromatous (BL), and lepromatous (LL). The borderline categories are immunologically unstable — the immune balance can shift toward either pole in response to changes in bacterial load, host immune status, pregnancy, concurrent infections, or the commencement of antileprosy treatment. These immune shifts manifest clinically as leprosy reactions: acute inflammatory episodes superimposed on the chronic disease that represent sudden changes in the immunological rheostat setting and that are the most common cause of acute nerve damage and disability in leprosy.
Type 1 reactions — reversal reactions — represent an upward shift toward the tuberculoid pole, with a sudden increase in cell-mediated immunity against M. leprae antigens producing acute inflammation within existing skin lesions and nerve trunks. Type 2 reactions — erythema nodosum leprosum — occur predominantly in lepromatous patients and represent immune complex-mediated inflammation driven by the high antigen load and high antibody titers characteristic of multibacillary disease. Both reaction types can cause acute, potentially permanent nerve damage within days to weeks, and their prompt recognition and treatment with corticosteroids or thalidomide is one of the most time-sensitive clinical decisions in leprosy management.
The Tuberculoid Pathway: Resistance at a Price
At the tuberculoid pole, the adaptive immune response is dominated by Th1 CD4-positive T lymphocytes that recognize mycobacterial antigens presented by macrophages and dendritic cells in the context of MHC class II molecules. These Th1 cells secrete the signature cytokines of the tuberculoid response: interleukin-2, which drives T-cell proliferation and sustains the clonal expansion of antigen-specific effector cells, and interferon-gamma, the quintessential macrophage-activating cytokine that induces the production of reactive oxygen and nitrogen intermediates, upregulates antigen presentation, and promotes the granulomatous cellular architecture that is the primary mechanism of mycobacterial containment.
The clinical consequence of this vigorous Th1 response is a disease that is bacteriologically contained but immunopathologically destructive. In tuberculoid leprosy, the mycobacterial burden is low — sometimes to the point where acid-fast bacilli cannot be detected even on multiple skin slit smears — and the risk of transmission is correspondingly reduced. But the intensely activated macrophages and lymphocytes that accomplish this bacterial suppression do so at the cost of collateral damage to the Schwann cells, axons, and connective tissue of the affected nerve. Nerve trunks in tuberculoid leprosy are thickened, tender, and surrounded by caseating or non-caseating granulomas that physically compress neural structures, disrupt blood supply, and ultimately destroy nerve fascicles with irreversible functional consequences.
Tuberculoid leprosy characteristically affects only one or a few nerve trunks and produces few, well-defined, hypopigmented skin lesions with clearly demarcated borders and complete loss of sensation within the lesion. This geographical containment reflects the localized and effective immunological barrier that the Th1 response constructs around the site of infection. The disease is self-limiting in many patients, capable of resolution without treatment in those with particularly robust cell-mediated immunity — though treatment remains essential to prevent progressive nerve damage during the active inflammatory phase.
The Lepromatous Pathway: Bacterial Triumph Through Immune Evasion
At the lepromatous pole, the immune balance has been tipped decisively in the bacterium's favor. The antigen-specific Th1 response is selectively anergic — patients with lepromatous leprosy fail to mount a delayed-type hypersensitivity reaction to intradermal lepromin, demonstrating that the specific immunological recognition of M. leprae antigens, rather than general immune competence, has been impaired. This anergy is actively maintained by a regulatory immune environment dominated by Th2 cells and regulatory T cells secreting immunosuppressive cytokines.
Interleukin-4, secreted by Th2 cells, redirects the immune response away from cell-mediated cytotoxicity and toward humoral immunity — a response that is relatively ineffective against an intracellular pathogen. Interleukin-10, produced by both Th2 cells and regulatory T cells, is a potent inhibitor of macrophage activation and Th1 cytokine production, directly antagonizing the interferon-gamma signaling that would otherwise activate mycobactericidal mechanisms. Transforming growth factor-beta, the signature cytokine of regulatory T-cell immunosuppression, further restrains the effector T-cell response and promotes tolerance to mycobacterial antigens. Together, these cytokines create an immunological environment in which M. leprae can proliferate essentially unchecked.
The cellular consequence of this permissive immune environment is the formation of Virchow cells — the foamy, lipid-laden macrophages that are the histopathological signature of lepromatous leprosy and the direct structural correlate of massive bacterial proliferation. Virchow cells are macrophages that have attempted and failed to kill internalized M. leprae, instead becoming overwhelmed with replicating bacteria that fill their cytoplasm with lipid-rich vacuoles. The lipid content of these vacuoles reflects both the bacterium's own lipid-rich cell wall and the accumulation of host cell lipids that M. leprae co-opts as energy substrates and membrane building materials. In lepromatous leprosy, Virchow cells accumulate in enormous numbers throughout the dermis, in peripheral nerve, in nasal mucosa, in the testes, and in other cooler body sites that provide the slightly below-body-temperature microenvironment that M. leprae preferentially inhabits.
The clinical picture of lepromatous leprosy reflects this bacteriologically permissive state. Skin lesions are numerous, poorly defined, symmetrically distributed, and filled with bacilli — each lesion a macroscopic manifestation of a site of uncontrolled mycobacterial replication. Peripheral nerve involvement is diffuse and bilateral, affecting multiple nerve trunks symmetrically in a stocking-and-glove distribution that contrasts sharply with the asymmetric, focal neuropathy of tuberculoid disease. The eyebrows, earlobes, and nasal bridge develop the characteristic leonine facies of advanced lepromatous disease as dermal macrophage infiltration thickens and distorts the facial skin. Nasal septal perforation, from direct mycobacterial destruction of the nasal mucosa and cartilage, can produce the saddle-nose deformity that is among the most historically recognizable stigmata of the disease.
The Ridley-Jopling Spectrum: Positions on the Dial
Between the tuberculoid and lepromatous poles lies a continuous immunological spectrum that the Ridley-Jopling classification system divides into five positions: tuberculoid (TT), borderline tuberculoid (BT), mid-borderline (BB), borderline lepromatous (BL), and lepromatous (LL). The borderline categories are immunologically unstable — the immune balance can shift toward either pole in response to changes in bacterial load, host immune status, pregnancy, concurrent infections, or the commencement of antileprosy treatment. These immune shifts manifest clinically as leprosy reactions: acute inflammatory episodes superimposed on the chronic disease that represent sudden changes in the immunological rheostat setting and that are the most common cause of acute nerve damage and disability in leprosy.
Type 1 reactions — reversal reactions — represent an upward shift toward the tuberculoid pole, with a sudden increase in cell-mediated immunity against M. leprae antigens producing acute inflammation within existing skin lesions and nerve trunks. Type 2 reactions — erythema nodosum leprosum — occur predominantly in lepromatous patients and represent immune complex-mediated inflammation driven by the high antigen load and high antibody titers characteristic of multibacillary disease. Both reaction types can cause acute, potentially permanent nerve damage within days to weeks, and their prompt recognition and treatment with corticosteroids or thalidomide is one of the most time-sensitive clinical decisions in leprosy management.
Diagnosis: Integrating Clinical, Bacteriological, and Histological Assessment
The diagnosis of leprosy remains fundamentally clinical in the field settings where the disease is most prevalent, guided by the WHO operational classification into paucibacillary and multibacillary disease based on lesion count and nerve involvement. The three cardinal signs — hypopigmented or erythematous skin lesions with impaired sensation, thickened peripheral nerves, and positive skin smear for acid-fast bacilli — together define the diagnosis with sufficient specificity for treatment initiation in resource-limited settings without access to histopathological or molecular confirmation.
In settings where histopathological evaluation is available, skin biopsy from the active edge of a lesion provides the definitive diagnostic confirmation and allows Ridley-Jopling classification based on the granuloma type, the lymphocytic infiltrate pattern, the degree of nerve involvement, and the bacillary index on modified Fite-Faraco staining for acid-fast organisms. Slit-skin smears from the earlobes, forehead, and any active lesion edge remain a rapid and inexpensive tool for establishing bacterial load and monitoring treatment response in multibacillary disease. Polymerase chain reaction for M. leprae-specific sequences — particularly the RLEP repetitive element and the folP1 gene used for drug resistance genotyping — is increasingly available as a molecular diagnostic and resistance surveillance tool.
Nerve conduction studies are a sensitive early indicator of peripheral nerve involvement, capable of detecting subclinical axonal and demyelinating changes before clinical sensory or motor deficits develop, and serial electrodiagnostic assessment is a valuable monitoring tool in patients with established nerve involvement. Ultrasound of peripheral nerve trunks — the ulnar at the elbow, the common peroneal at the fibular head, the great auricular at the neck — reveals the nerve thickening and heterogeneity of echotexture that are sonographically characteristic of leprous neuritis and can guide nerve biopsy when the diagnosis remains in question.
In settings where histopathological evaluation is available, skin biopsy from the active edge of a lesion provides the definitive diagnostic confirmation and allows Ridley-Jopling classification based on the granuloma type, the lymphocytic infiltrate pattern, the degree of nerve involvement, and the bacillary index on modified Fite-Faraco staining for acid-fast organisms. Slit-skin smears from the earlobes, forehead, and any active lesion edge remain a rapid and inexpensive tool for establishing bacterial load and monitoring treatment response in multibacillary disease. Polymerase chain reaction for M. leprae-specific sequences — particularly the RLEP repetitive element and the folP1 gene used for drug resistance genotyping — is increasingly available as a molecular diagnostic and resistance surveillance tool.
Nerve conduction studies are a sensitive early indicator of peripheral nerve involvement, capable of detecting subclinical axonal and demyelinating changes before clinical sensory or motor deficits develop, and serial electrodiagnostic assessment is a valuable monitoring tool in patients with established nerve involvement. Ultrasound of peripheral nerve trunks — the ulnar at the elbow, the common peroneal at the fibular head, the great auricular at the neck — reveals the nerve thickening and heterogeneity of echotexture that are sonographically characteristic of leprous neuritis and can guide nerve biopsy when the diagnosis remains in question.
Treatment: Multidrug Therapy and Beyond
The introduction of WHO-recommended multidrug therapy in 1982 — combining rifampicin, dapsone, and clofazimine in standard six- or twelve-month regimens for paucibacillary and multibacillary disease respectively — transformed leprosy from a lifelong, stigmatizing, progressively disabling condition into a curable disease. The global leprosy burden has fallen by more than ninety-five percent since the widespread implementation of multidrug therapy, and the WHO's sustained commitment to providing free multidrug therapy to all registered leprosy patients globally represents one of the most successful public health interventions in the history of infectious disease control.
The management of leprosy reactions requires immunosuppressive therapy administered alongside, not instead of, antileprosy chemotherapy. High-dose prednisolone is the first-line treatment for both Type 1 reversal reactions and severe erythema nodosum leprosum, with thalidomide providing additional efficacy for refractory or recurrent Type 2 reactions in male patients and postmenopausal women — its teratogenicity precluding its use in women of childbearing potential. The corticosteroid regimen for reversal reactions typically extends over six months or longer, reflecting the sustained immunological instability of borderline patients whose immune rheostat is being reset by antimycobacterial therapy.
The management of established nerve damage and disability — the consequence of delayed diagnosis, untreated reactions, or inadequate corticosteroid coverage — falls to the domains of physiotherapy, occupational therapy, reconstructive surgery, and community-based rehabilitation. Tendon transfer procedures can restore function to the claw hand and drop foot of advanced leprosy neuropathy; tarsal tunnel release can prevent further ischemic nerve injury in patients with nerve trunk compression; plantar pressure-offloading footwear substantially reduces the incidence of neuropathic ulceration. These interventions do not reverse the nerve damage that M. leprae has inflicted, but they mitigate its functional consequences — and they remind us that the management of leprosy is not complete when the bacterium is cleared but continues throughout the lifetime of the nerve damage it leaves behind.
The management of leprosy reactions requires immunosuppressive therapy administered alongside, not instead of, antileprosy chemotherapy. High-dose prednisolone is the first-line treatment for both Type 1 reversal reactions and severe erythema nodosum leprosum, with thalidomide providing additional efficacy for refractory or recurrent Type 2 reactions in male patients and postmenopausal women — its teratogenicity precluding its use in women of childbearing potential. The corticosteroid regimen for reversal reactions typically extends over six months or longer, reflecting the sustained immunological instability of borderline patients whose immune rheostat is being reset by antimycobacterial therapy.
The management of established nerve damage and disability — the consequence of delayed diagnosis, untreated reactions, or inadequate corticosteroid coverage — falls to the domains of physiotherapy, occupational therapy, reconstructive surgery, and community-based rehabilitation. Tendon transfer procedures can restore function to the claw hand and drop foot of advanced leprosy neuropathy; tarsal tunnel release can prevent further ischemic nerve injury in patients with nerve trunk compression; plantar pressure-offloading footwear substantially reduces the incidence of neuropathic ulceration. These interventions do not reverse the nerve damage that M. leprae has inflicted, but they mitigate its functional consequences — and they remind us that the management of leprosy is not complete when the bacterium is cleared but continues throughout the lifetime of the nerve damage it leaves behind.
Conclusion: Three Phases, One Extraordinary Pathogen
The three-phase framework presented in Figure 1 distills decades of cellular and molecular leprosy research into a coherent pathogenetic narrative. Phase 1 reveals a bacterium that has evolved a molecular key — PGL-1 — perfectly shaped for the lock of alpha-dystroglycan on the Schwann cell surface, achieving intracellular entry not through force but through biochemical deception. Phase 2 reveals a bacterium that, once inside, does not merely shelter within its host cell but actively transforms it — suppressing the transcriptional program of the mature Schwann cell, inducing demyelination and dedifferentiation, and establishing a stem-cell-like intracellular environment optimized for bacterial persistence and dissemination. Phase 3 reveals that the ultimate outcome of this molecular hijacking depends not on the bacterium alone but on the immunological character of the host — a rheostat whose Th1-dominated tuberculoid setting offers bacterial resistance at the cost of nerve destruction, and whose Th2/T-reg-dominated lepromatous setting permits bacterial proliferation at the cost of widespread tissue infiltration.
What makes Morbus Hansen scientifically extraordinary is not its prevalence — it is, by global standards, a relatively rare disease — but the precision and sophistication of the molecular mechanisms through which a metabolically minimal organism with the slowest doubling time of any human pathogen manages to establish itself in one of the most immunologically privileged tissues of the human body, reprogram that tissue's cellular identity, and then exploit the host's own immune response as a determinant of its ultimate fate. Each of these phases, studied in sufficient molecular detail, reveals biology that extends far beyond leprosy itself: principles of mycobacterial pathogenesis, of peripheral glial biology, of T-helper cell polarization, and of neural crest stem cell plasticity that illuminate far broader questions in immunology and neurobiology.
For the clinician encountering leprosy at the bedside, this mechanistic understanding translates into a more informed recognition of what is at stake at each phase of the disease: the critical window of early diagnosis before irreversible nerve damage accumulates, the importance of immune monitoring to detect and treat reactions before they scar peripheral nerves beyond recovery, and the recognition that the skin lesion visible to the eye is the surface expression of a molecular drama that has been playing out, invisibly, within the peripheral nervous system from the moment of first bacterial entry. In a disease whose physical stigmata have defined its social history for millennia, it is fitting that the deepest understanding of its pathogenesis should be found not on the surface of the skin, but within the cell that the bacterium has chosen as its home.
What makes Morbus Hansen scientifically extraordinary is not its prevalence — it is, by global standards, a relatively rare disease — but the precision and sophistication of the molecular mechanisms through which a metabolically minimal organism with the slowest doubling time of any human pathogen manages to establish itself in one of the most immunologically privileged tissues of the human body, reprogram that tissue's cellular identity, and then exploit the host's own immune response as a determinant of its ultimate fate. Each of these phases, studied in sufficient molecular detail, reveals biology that extends far beyond leprosy itself: principles of mycobacterial pathogenesis, of peripheral glial biology, of T-helper cell polarization, and of neural crest stem cell plasticity that illuminate far broader questions in immunology and neurobiology.
For the clinician encountering leprosy at the bedside, this mechanistic understanding translates into a more informed recognition of what is at stake at each phase of the disease: the critical window of early diagnosis before irreversible nerve damage accumulates, the importance of immune monitoring to detect and treat reactions before they scar peripheral nerves beyond recovery, and the recognition that the skin lesion visible to the eye is the surface expression of a molecular drama that has been playing out, invisibly, within the peripheral nervous system from the moment of first bacterial entry. In a disease whose physical stigmata have defined its social history for millennia, it is fitting that the deepest understanding of its pathogenesis should be found not on the surface of the skin, but within the cell that the bacterium has chosen as its home.
Urinary Tract Infection Updates: Current Clinical and Public Health Developments
Urinary tract infection remains one of the most common bacterial infections globally, with current updates focusing on antimicrobial resistance, diagnostic stewardship, recurrent infection management, catheter-associated prevention, and emerging non-antibiotic strategies. This article reviews recent developments relevant to clinicians, researchers, and public health professionals.
Top 5 Vegetables That May Enhance Immune Function: An Evidence-Based Nutritional Overview
Dietary patterns rich in vegetables are consistently associated with improved health outcomes, including support of normal immune function. This article reviews five vegetables—broccoli, spinach, garlic, carrots, and red bell peppers—that may contribute to immune resilience through their content of vitamins, minerals, antioxidants, and bioactive phytochemicals.


Understanding Stunted Children: Chronic Growth Failure, Early-Life Risks, and Why It Matters Beyond Height
Stunting in children refers to impaired linear growth resulting from chronic undernutrition, repeated infection, and unfavorable early-life conditions. More than a matter of short stature, stunting reflects a broader process of biological and developmental disadvantage that can affect cognitive outcomes, school performance, and long-term health.
Blood Pressure Monitoring as a Public Health Priority: Strengthening Early Detection and Long-Term Cardiovascular Prevention
Blood pressure monitoring remains one of the most practical and impactful tools in public health for identifying hypertension early, guiding treatment decisions, and reducing long-term cardiovascular risk. Wider adoption of accurate office, community, and home-based monitoring strategies could significantly improve prevention of stroke, heart disease, kidney damage, and premature mortality.

